This is probably one of those “solution looking for a problem” ideas. I seem to have a lot of those! Anyway, here’s the idea. This tool would allow you to set up a sequence of timed events. Each event has a title, a duration, and a sound to play when the event starts. You can also set the text and background colours for the events. All the events are played in sequence from start to finish. You’d create a timer sequence by dragging new events onto a kind of timeline, and you could drag them around to rearrange them, and edit their properties.
One possible use for this is as a workout timer. If you have a bunch of exercises you want to perform for different lengths of time, you could set up a sequence like this:
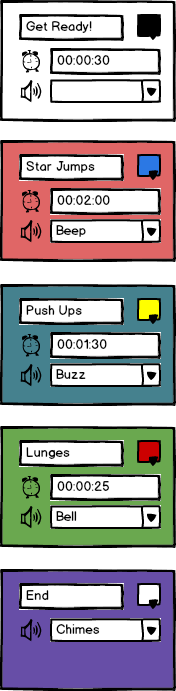 I haven’t shown this, but there would be some way of adding new events, deleting them and dragging them around to change the order. You could also give your sequences names and save them for later, or even share them. When you run the sequence, each event would run full-screen, playing the appropriate sound. So for the 3rd event in the above sequence, you’d hear a buzz sound at the start, and see something like this:
I haven’t shown this, but there would be some way of adding new events, deleting them and dragging them around to change the order. You could also give your sequences names and save them for later, or even share them. When you run the sequence, each event would run full-screen, playing the appropriate sound. So for the 3rd event in the above sequence, you’d hear a buzz sound at the start, and see something like this:
 The final event is a bit different because it doesn’t have a duration, so it just stays there until you close it.
The final event is a bit different because it doesn’t have a duration, so it just stays there until you close it.
I can think of a few possible uses for this tool. A workout timer as shown above is one of them. It might also be useful as a kitchen timer if you’re attempting to cook several different things and have them ready at the same time (as long as you plan ahead and work out all the steps you need to do in advance). If you use the Pomodoro Technique, this could be used as a timer for that, with 25 minute work sessions, and 5 minute breaks all set up as events in advance.
Another way to think of this tool is as a way of creating very simple automated presentations or slideshows. It could allow you to set a background image for each event instead of a plain colour. Then you could create timed slideshows and presentations really easily, which opens up a lot more possible uses.